High Blood Pressure in Adrenal Gland Cancer (Pheochromocytoma)
High Blood Pressure in Adrenal Gland Cancer (Pheochromocytoma): Causes, Symptoms, and Management
- High Blood Pressure in Adrenal Gland Cancer (Pheochromocytoma): Causes, Symptoms, and Management
- Why High Blood Pressure Can Occur in Adrenal Gland Cancer (Pheochromocytoma)
- The Hormonal Mechanism Behind Tumor-Induced Hypertension
- How Common Is High Blood Pressure in Patients with Adrenal Tumors?
- What Triggers This Symptom in Cancer Patients?
- When to Seek Immediate Medical Help for Tumor-Related Hypertension
- Diagnostic Approach: How Doctors Confirm the Cause
- Treatment Strategies: Controlling Hypertension and Tumor Activity
- Can You Prevent High Blood Pressure in Adrenal Gland Cancer?
- Will Blood Pressure Return to Normal After Tumor Treatment?
- What Doctors Say About Treating Hypertension in Adrenal Cancer
- Questions to Ask Your Doctor
- FAQ: High Blood Pressure and Pheochromocytoma
Why High Blood Pressure Can Occur in Adrenal Gland Cancer (Pheochromocytoma)
High blood pressure, or hypertension, is often one of the earliest and most dangerous signs of adrenal gland tumors—especially pheochromocytoma. This rare tumor develops in the adrenal medulla and secretes excess catecholamines, such as epinephrine and norepinephrine. These hormones are responsible for the body’s “fight or flight” response, and when produced uncontrollably by tumor cells, they cause sustained or episodic spikes in blood pressure.
Unlike typical hypertension, which is often gradual and related to lifestyle, the hypertension seen in adrenal tumors is abrupt, severe, and resistant to common treatments. Patients may experience pounding headaches, palpitations, sweating, and anxiety during these episodes. These symptoms often mimic panic attacks but stem from an endocrine imbalance.
Many people are unaware of this link until they are diagnosed with an adrenal tumor. In cases involving Gland Cancer, particularly of the adrenal glands, recognizing the pattern of hormonal hypertension is crucial for early intervention.
The Hormonal Mechanism Behind Tumor-Induced Hypertension
The adrenal medulla is responsible for producing catecholamines—specifically adrenaline (epinephrine), noradrenaline (norepinephrine), and in some cases, dopamine. Pheochromocytoma arises from chromaffin cells in this region and leads to excess secretion of these hormones into the bloodstream.
This hormonal surge triggers a cascade of effects: vasoconstriction, increased cardiac output, elevated blood sugar, and stimulated sympathetic nervous activity. The most immediate result is a dramatic rise in blood pressure, which may fluctuate unpredictably or persist at dangerously high levels.
In some cases, the tumor may also disrupt other hormonal systems, including androstenedione metabolism, affecting additional endocrine functions such as libido, fertility, and body hair regulation. While these effects are secondary, they may serve as clues when diagnosing complex adrenal pathology.
How Common Is High Blood Pressure in Patients with Adrenal Tumors?
Although pheochromocytoma is rare—occurring in about 2 to 8 people per million annually—it is present in roughly 0.1–0.6% of all patients with hypertension. Among those with adrenal gland tumors, hypertension is a leading symptom.
Clinical Incidence of Hypertension in Pheochromocytoma
| Patient Group | Prevalence of Hypertension |
| Patients with diagnosed pheochromocytoma | 90–95% |
| Individuals with adrenal incidentaloma | ~10% show hormonal activity leading to hypertension |
| Pediatric patients with adrenal tumors | ~70–85% |
| Genetic syndromes (e.g., MEN2, VHL) | Up to 100% in active tumors |
In hereditary syndromes, the detection of high blood pressure often leads to further screening and early diagnosis of pheochromocytoma. This is especially important in cases where thyroid cancer or other endocrine neoplasms coexist, as seen in multiple endocrine neoplasia (MEN) syndromes.
What Triggers This Symptom in Cancer Patients?
The causes of high blood pressure in adrenal gland cancer are both direct and systemic. The primary trigger is catecholamine overproduction, but additional contributors may include tumor size, vascular involvement, and stress responses to malignancy itself.
Common Mechanisms Leading to Hypertension
| Cause Type | Mechanism Description |
| Tumor Secretion | Overproduction of epinephrine and norepinephrine by chromaffin cells |
| Vascular Compression | Large tumors may compress renal arteries, mimicking renal hypertension |
| Medication Effects | Chemotherapy or corticosteroids may exacerbate blood pressure spikes |
| Surgical Stress | Tumor manipulation during biopsy or surgery can provoke a hypertensive crisis |
| Paraneoplastic Syndromes | Rare endocrine interactions can cause secondary hormonal imbalances |
Even after tumor removal, residual hypertension may persist for weeks to months, depending on the tumor’s size, duration of hormone exposure, and organ damage incurred during the active disease phase.
When to Seek Immediate Medical Help for Tumor-Related Hypertension
Hypertension caused by adrenal tumors like pheochromocytoma is often volatile and may progress rapidly to life-threatening crises if untreated. While some patients report mild symptoms initially, others present with sudden hypertensive emergencies that demand urgent care.
Critical warning signs include severe, throbbing headaches, chest pain, vision disturbances, rapid heart rate, and profuse sweating—particularly if they occur in episodes. These “spells” may be mistaken for panic attacks or cardiac events but are actually the hallmark of a catecholamine surge.
Hypertensive crisis can lead to stroke, heart attack, or organ failure if not promptly managed. If a patient with known or suspected gland cancer experiences these symptoms, especially in conjunction with palpitations or anxiety, they should seek emergency evaluation without delay.
In surgical settings, manipulating the tumor—during biopsy or resection—can provoke a massive hormone release, which makes preoperative management and stabilization absolutely critical.
Diagnostic Approach: How Doctors Confirm the Cause
Accurate diagnosis of hypertension caused by pheochromocytoma involves a combination of hormonal assays and imaging. Since symptoms can be intermittent, capturing biochemical evidence during or shortly after a hypertensive episode is ideal.
Key Diagnostic Tools and Their Purpose
| Test/Procedure | Role in Diagnosis |
| Plasma Free Metanephrines | Sensitive test for detecting catecholamine excess |
| 24-hour Urine Catecholamines | Confirms hormone overproduction over time |
| Abdominal CT or MRI | Localizes adrenal tumor and estimates size |
| MIBG Scintigraphy | Detects active catecholamine-producing tissue |
| Genetic Testing | Recommended in familial cases or bilateral tumors |
Plasma metanephrines are now the gold standard due to their high sensitivity. However, results must be interpreted carefully, especially in patients under stress or on interfering medications.
When the tumor is suspected but not visualized on standard imaging, MIBG or PET scans help detect smaller or extra-adrenal lesions. Importantly, physicians often screen for coexisting tumors like thyroid cancer in syndromic cases.
Treatment Strategies: Controlling Hypertension and Tumor Activity
Managing blood pressure in adrenal gland cancer involves two parallel goals: stabilize the patient’s cardiovascular system and control the hormone-secreting tumor.
Integrated Treatment Plan
| Treatment Component | Clinical Role |
| Alpha-blockers (e.g., phenoxybenzamine) | First-line therapy to block vasoconstriction |
| Beta-blockers | Added after alpha-blockade to control heart rate |
| Calcium Channel Blockers | Alternative or adjunct agents for blood pressure control |
| Surgical Tumor Resection | Definitive treatment, often curative in localized tumors |
| Preoperative Optimization | Mandatory to prevent intraoperative hypertensive crisis |
| Chemotherapy or Radiotherapy | Used in metastatic or inoperable cases |
Patients must be started on alpha-blockers at least 10–14 days before surgery. Beta-blockers are never used first because unopposed alpha stimulation can worsen blood pressure.
For inoperable tumors or those with metastasis, targeted therapies and chemotherapy may be used to slow progression. In such cases, long-term medical management of blood pressure remains essential.
Can You Prevent High Blood Pressure in Adrenal Gland Cancer?
While it’s not always possible to prevent tumor-induced hypertension, early recognition and monitoring in high-risk individuals can reduce complications significantly. Patients with a family history of pheochromocytoma or those with known endocrine syndromes (e.g., MEN2, VHL) benefit from genetic screening and regular biochemical tests.
Lifestyle factors—though not primary causes—can influence severity. High-stress levels, stimulant use (caffeine, decongestants), and certain drugs can trigger hypertensive episodes in those with latent tumors.
Preventive Measures for High-Risk Individuals
| Measure | Benefit |
| Annual screening (plasma metanephrines) | Early tumor detection |
| Genetic counseling/testing | Identifies inherited risk |
| Avoidance of stimulants | Reduces risk of triggering a hypertensive spell |
| Prophylactic adrenal imaging | Useful in syndromic families |
| Pre-treatment monitoring in cancer patients | Identifies secondary hypertension early |
In patients already diagnosed with androstenedione imbalance or other adrenal abnormalities, vigilant follow-up is essential. Multidisciplinary care involving endocrinologists, cardiologists, and oncologists provides the most effective prevention and intervention strategy.
Will Blood Pressure Return to Normal After Tumor Treatment?
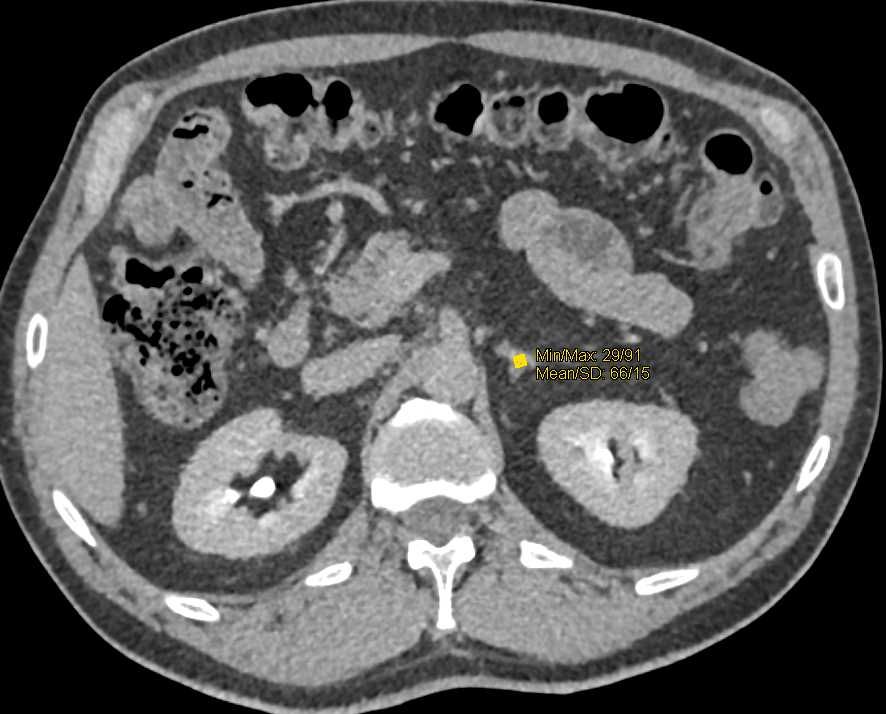
In many patients, successful treatment of pheochromocytoma leads to significant improvement or complete resolution of hypertension. After surgical resection, blood pressure often normalizes within days to weeks as catecholamine levels stabilize.
However, the outcome depends on multiple factors: the duration and severity of preoperative hypertension, the presence of organ damage (e.g., cardiac hypertrophy or renal impairment), and whether the tumor was completely removed. Patients with long-standing disease may have residual hypertension due to vascular remodeling or secondary kidney dysfunction.
Post-Treatment Prognosis Based on Clinical Factors
| Clinical Scenario | Blood Pressure Outlook |
| Small, localized tumor, early detection | Full normalization likely |
| Large tumor with cardiovascular damage | Partial improvement; may need medication |
| Bilateral or metastatic disease | Lifelong hypertension management likely |
| Familial pheochromocytoma (MEN, VHL) | Ongoing risk of recurrence; regular monitoring needed |
While some cases result in permanent cure, others require long-term antihypertensive therapy even after tumor removal. Therefore, continuous follow-up with endocrinologists and cardiologists is vital.
What Doctors Say About Treating Hypertension in Adrenal Cancer
Endocrinologists and oncologists emphasize that hypertension in adrenal tumors is both a diagnostic clue and a therapeutic challenge. Many patients are misdiagnosed with essential hypertension until further investigation reveals an underlying tumor.
Doctors stress the importance of distinguishing pheochromocytoma-related hypertension from other forms. Inappropriate use of medications—such as prescribing beta-blockers first—can lead to hypertensive crisis in this setting. Clinicians also note that symptoms like episodic palpitations, anxiety, and sweating should always raise suspicion in younger hypertensive patients.
According to oncologists specializing in gland cancer, multidisciplinary care yields the best outcomes. Surgery, anesthesia, endocrinology, and oncology teams must coordinate closely—especially in high-risk operations. Early alpha-blockade and comprehensive preoperative planning dramatically reduce complications and improve long-term survival.
Questions to Ask Your Doctor
Being informed empowers patients and families to make safer, faster, and more confident decisions. Here are questions that can guide meaningful discussions with your care team:
| Key Questions | Why It Matters |
| What type of adrenal tumor do I have? | Confirms whether it’s hormonally active |
| How do you know this tumor is causing my blood pressure issues? | Links symptoms with diagnosis |
| Do I need genetic testing for inherited syndromes? | Identifies long-term and familial risk |
| What is the best imaging for tumor localization? | Helps decide next diagnostic steps |
| What medications will I need before surgery? | Ensures safe preparation for tumor resection |
| Can surgery completely cure the hypertension? | Sets expectations |
| What are the risks of untreated pheochromocytoma? | Clarifies urgency |
| Will I need hormone therapy after surgery? | Addresses long-term endocrine balance |
| Can this tumor spread or come back? | Explores recurrence risk |
| What symptoms should prompt emergency care? | Prepares for hypertensive crises |
| Are there dietary or lifestyle triggers I should avoid? | Prevents symptom flares |
| Should I avoid certain medications or activities? | Reduces crisis risk |
| How often will I need follow-up tests? | Sets monitoring schedule |
| Will you coordinate care with an endocrinologist? | Improves outcomes |
| Could this condition be related to other endocrine issues like thyroid cancer? | Identifies syndromic conditions |
FAQ: High Blood Pressure and Pheochromocytoma
1. What makes hypertension from pheochromocytoma different from regular high blood pressure?
It’s caused by excessive adrenaline production, leading to sudden spikes rather than gradual elevation. It’s often resistant to common medications.
2. Can I have this tumor without symptoms?
Yes. Some adrenal tumors are “silent” but still release hormones periodically or under stress.
3. Is pheochromocytoma always cancerous?
No. Most are benign, but about 10% can be malignant. Cancerous forms may spread to lymph nodes, liver, lungs, or bone.
4. How is the tumor usually found?
Through symptoms, biochemical tests, and imaging. Sometimes incidentally during scans for unrelated conditions.
5. Is hypertension the first symptom in most patients?
Often, yes. Especially if it appears suddenly in a younger person or is hard to control with standard medications.
6. What’s the treatment for the tumor itself?
Surgical removal is the mainstay. Some may also need radiation or chemotherapy if the tumor is malignant.
7. What if I can’t have surgery?
Medical therapy with alpha-blockers and supportive care may control symptoms, especially if the tumor is inoperable.
8. Can pregnancy trigger pheochromocytoma symptoms?
Yes, pregnancy can worsen symptoms or unmask the tumor due to hormonal shifts. Diagnosis is often delayed in these cases.
9. Will my children inherit this condition?
Possibly. About 30–40% of pheochromocytomas are linked to genetic syndromes. Genetic counseling is advised.
10. Can this tumor affect other hormones?
Yes. Some affect androstenedione, cortisol, or even sex hormones depending on their location and structure.
11. Is it safe to exercise with this tumor?
Light activity may be safe under medical supervision, but strenuous exertion can trigger hormone release.
12. Can stress make my symptoms worse?
Absolutely. Emotional or physical stress can elevate catecholamines and worsen hypertension.
13. What are common medication mistakes to avoid?
Using beta-blockers before alpha-blockers, or taking stimulant drugs like pseudoephedrine, can provoke a crisis.
14. Will I need lifelong medication after treatment?
Not always. If cured surgically and without complications, medications may be tapered or stopped.
15. Is follow-up important after recovery?
Yes. Lifelong monitoring is essential, especially in genetic cases or if the tumor was malignant.